
Оглавление
Разработка терапевтических антител — сложный процесс, требующий оптимизации множества биофизических свойств: высокой экспрессии, стабильности и низкой склонности к агрегации. В отличие от простых точечных мутаций в белках, оптимизация антител особенно сложна: сочетание VH (вариабельной тяжелой) и VL (вариабельной легкой) доменов создает огромное комбинаторное пространство, где даже небольшие изменения могут кардинально повлиять на фолдинг, связывание или пригодность к разработке.
Экспериментальные измерения и машинное обучение
Экспериментальное предсказание этих эффектов дорого и требует много времени, поэтому машинное обучение на данных последовательностей антител стало важным инструментом. Обучающие представления последовательностей антител, языковые модели белков могут захватывать отношения последовательность-структура-функция, которые помогают направлять выбор дизайна.
Набор данных GDPa1
Набор данных GDPa1 предоставляет парные последовательности антител VH/VL с экспериментально измеренными тестами на пригодность к разработке, включая выход экспрессии, гидрофобность, стабильность и само-взаимодействие.
Это делает его ценным эталоном для проверки того, могут ли языковые модели белков, такие как p-IgGen, генерировать эмбеддинги, которые обобщаются по различным тестам и предсказывают результаты пригодности к разработке.
В разделах ниже мы демонстрируем, как:
- Создавать эмбеддинги пар VH/VL с помощью p-IgGen
- Обучать простые регрессионные модели на различных целевых тестах
- Оценивать обобщение с помощью перекрестной проверки с учетом кластеров и изотипов
Быстрый обзор
Последовательности антител: VH и VL
Антитела — это Y-образные белки, состоящие из двух цепей:
- VH (Variable Heavy): вариабельная область тяжелой цепи, которая участвует в связывании антигена
- VL (Variable Light): вариабельная область легкой цепи, которая соединяется с VH для формирования полного сайта связывания
Вместе VH и VL кодируют аминокислотные последовательности, которые определяют специфичность и биохимические свойства антитела.
В этом наборе данных каждая строка включает обе последовательности VH и VL для данного антитела.
Доступные свойства
Каждое антитело имеет связанные экспериментальные измерения по 5 тестам (не все измерения доступны для каждой последовательности):
- Titer: Выход экспрессии антитела в клетках млекопитающих
- HIC (Hydrophobic Interaction Chromatography): Показатель гидрофобности и склонности к агрегации
- PR_CHO: Полиреактивность в клетках CHO (китайского хомячка) — измеряет, насколько антитело связывается с другими белками, с которыми оно не должно связываться
- Tm2: Термическая стабильность (температура плавления домена CH2)
- AC-SINS_pH7.4: Склонность к само-взаимодействию (более высокие значения часто коррелируют с плохой пригодностью к разработке)
Примечание о влиянии изотипов
Антитела в этом наборе данных GDPa1 имеют разные подклассы IgG (IgG1, IgG2, IgG4). Некоторые измерения, такие как термическая стабильность (Tm2), сильно зависят от изотипа антитела. Разные подклассы IgG (например, IgG1, IgG2, IgG4) имеют систематические различия в стабильности домена CH2, которые могут затмевать тонкие вариации на уровне последовательности — поэтому рассмотрите возможность включения подкласса в качестве признака в ваших моделях! Чтобы проиллюстрировать это, мы можем построить простую boxplot диаграмму значений `Tm2`, сгруппированных по изотипу.
plt.figure(figsize=(6,4))
sns.boxplot(data=df, x="hc_subtype", y="Tm2")
plt.title("Distribution of Tm2 scores by antibody isotype")
plt.xlabel("Isotype")
plt.ylabel("Tm2 (°C)")
plt.show()
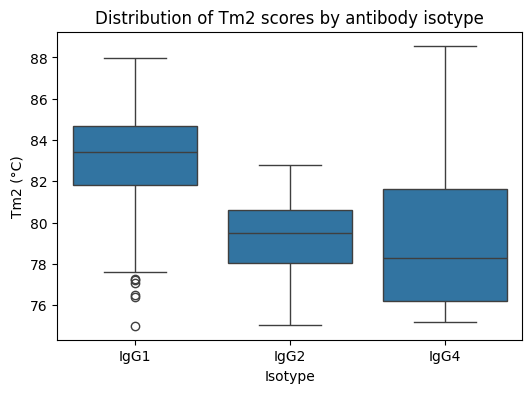
Ирония ситуации в том, что даже самые продвинутые языковые модели для белков сталкиваются с фундаментальной проблемой биоинформатики: изотип антитела оказывает большее влияние на стабильность, чем тонкие вариации последовательности. Это напоминает, что машинное обучение в биологии — это не просто техническая задача, а постоянный диалог между алгоритмами и биологической реальностью, где иногда простой категориальный признак важнее сложных эмбеддингов.
В этом руководстве мы выбираем одну цель за раз (например, `HIC`) и обучаем простую линейную модель на объединенных эмбеддингах последовательностей для оценки предсказательной способности.
Мы начинаем с импорта основных библиотек для обработки данных, визуализации, статистики и моделирования. Используя Hugging Face Datasets, мы загружаем набор данных GDPa1 в Pandas DataFrame, проверяем доступные столбцы тестов и проверяем наличие пропущенных значений. Для этого руководства мы выбираем одну цель пригодности к разработке (здесь `»HIC»`, хотя другие, такие как `»Titer»`, также могут быть выбраны) и удаляем строки с пропущенными измерениями, чтобы набор данных был готов для обучения и оценки модели.
from datasets import load_dataset import matplotlib.pyplot as plt import numpy as np from scipy.stats import spearmanr import seaborn as sns from sklearn.linear_model import Ridge from sklearn.model_selection import train_test_split import torch from tqdm.auto import tqdm from transformers import AutoModelForCausalLM, AutoTokenizer
model_name = "ollieturnbull/p-IgGen"
df = load_dataset("ginkgo-datapoints/GDPa1")["train"].to_pandas()
# Показать количество NaN по тестам
print(df[["Titer", "HIC", "PR_CHO", "Tm2", 'AC-SINS_pH7.4']].isna().sum())
target = "HIC"
# Пример: Просто предсказываем HIC, поэтому удалим строки с NaN для этого
df = df.dropna(subset=[target])
# output Titer 7 HIC 4 PR_CHO 49 Tm2 53 AC-SINS_pH7.4 4
Белковые последовательности VH и VL для каждого антитела объединяются в единую токенизированную строку ввода для p-IgGen. Мы добавляем токен `»1″` в начале и токен `»2″` в конце, чтобы обозначить границы последовательности.
# Tokenize the sequences
tokenizer = AutoTokenizer.from_pretrained(model_name)
# Paired sequence handling: Concatenate heavy and light chains and add beginning ("1") and end ("2") tokens
# (e.g. ["EVQLV...", "DIQMT..."] -> "1E V Q L V ... D I Q M T ... 2")
sequences = [\
"1" + " ".join(heavy) + " ".join(light) + "2"\
for heavy, light in zip(\
df["vh_protein_sequence"],\
df["vl_protein_sequence"],\
)\
]
print(sequences[0])
# output 1Q V K L Q E S G A E L A R P G A S V K L S C K A S G Y T F T N Y W M Q W V K Q R P G Q G L D W I G A I Y P G D G N T R Y T H K F K G K A T L T A D K S S S T A Y M Q L S S L A S E D S G V Y Y C A R G E G N Y A W F A Y W G Q G T T V T V S SD I E L T Q S P A S L S A S V G E T V T I T C Q A S E N I Y S Y L A W H Q Q K Q G K S P Q L L V Y N A K T L A G G V S S R F S G S G S G T H F S L K I K S L Q P E D F G I Y Y C Q H H Y G I L P T F G G G T K L E I K2
Мы загружаем модель p-IgGen и передаем батчи токенизированных последовательностей VH/VL для получения эмбеддингов скрытых состояний. Для каждой последовательности мы вычисляем усредненное представление по токенам, получая вектор фиксированной длины, который захватывает информацию о последовательности на уровне антитела.
# Load model
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
model = AutoModelForCausalLM.from_pretrained(model_name).to(device)
# Takes about 60 seconds for 242 sequences on my CPU, and 1.1s on GPU
batch_size = 16
mean_pooled_embeddings = []
for i in tqdm(range(0, len(sequences), batch_size)):
batch = tokenizer(sequences[i:i+batch_size], return_tensors="pt", padding=True, truncation=True)
outputs = model(batch["input_ids"].to(device), return_rep_layers=[-1], output_hidden_states=True)
embeddings = outputs["hidden_states"][-1].detach().cpu().numpy()
mean_pooled_embeddings.append(embeddings.mean(axis=1))
mean_pooled_embeddings = np.concatenate(mean_pooled_embeddings)
С эмбеддингами в качестве признаков (`X`) и выбранным измерением теста в качестве цели (`y`) мы разделяем данные на обучающую и тестовую выборки. По сообщению Hugging Face, этот подход демонстрирует, как современные языковые модели могут ускорить разработку терапевтических антител.





Оставить комментарий